PhyML 3.0
Overview: new algorithms, methods and utilities PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient…

FastME 2.0
FastME is a software package for the fast and accurate inference of phylogenetic trees from distance matrices. It implements algorithms based on the Balanced Minimum Evolution (BME) principle, a distance-based criterion closely related to the Neighbor Joining (NJ) method. The goal of the BME framework is to identify the phylogenetic tree that minimizes the total…

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…
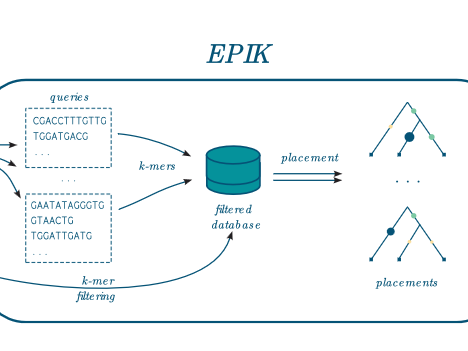
EPIK
Precise and scalable evolutionary placement with informative k-mers EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and…

TFscope
Characterizing the binding preferences of transcription factors (TFs) in different cell types and conditions is key to understand how they orchestrate gene expression. TFscope is a machine learning approach that identifies sequence features explaining the binding differences observed between two ChIP-seq experiments targeting either the same TF in two conditions or two TFs with similar…

AQUAPONY
AquaPony: interactive visualization of phylogeographic scenarios AquaPony is a web application designed to explore and interpret evolutionary scenarios on annotated phylogenetic trees (for example, ancestral geographic states). It was built to make uncertainty in ancestral reconstructions easier to understand and communicate. Why AquaPony? In phylogeography, several scenarios can be nearly as plausible as the best…

DExTER
Overview DExTER (Domain Exploration To Explain gene Regulation) is a bioinformatics tool designed to automatically identify genomic regions whose nucleotide composition correlates with gene expression levels. Unlike traditional approaches focusing on short transcription factor binding sites (6-12 bp), DExTER detects Long Regulatory Elements (LREs) that can span tens to hundreds of nucleotides. This makes it…

CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared…

TIDE
What is TIDE? TIDE is the orchestration platform behind ATGC online bioinformatics services. It provides a unified web interface and a public REST API to submit analyses, monitor jobs, and retrieve results across a growing catalog of phylogenetic and sequence-analysis tools. TIDE is a modernized redesign of WAVES, an older tool previously developped by ATGC…