PhyML 3.0
Overview: new algorithms, methods and utilities
PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient options to search the space of phylogenetic tree topologies.
Installation
To install PhyML, download the code from https://github.com/stephaneguindon/phyml/. After unpacking the archive, go into the phyml/ folder and type the following command:
sh ./autogen.sh;If you are using a Mac computer or running a Unix-like operating system, you will need to install the packages autoconf automake and pkg-config. On a Mac, the following command should set you up (provided Homebrew is installed on your Mac…): brew install pkg-config autoconf automake;
Next, to install any program that is part of the PhyML package, type the following commands:
./configure --enable-phyml;
make;To compile a Windows executable, install MinGW and run:
./configure --enable-win --enable-phyml;
make;To install the MPI version of PhyML, type the following commands:
autoreconf -i;
./configure --enable-phyml-mpi;
make;PhyML 3.0 online execution
Other tools

WAVES
Summary WAVES is a web application dedicated to bioinformatic tool integration. It provides an efficient way to implement a service for any bioinformatic software. Such services are automatically made available in three ways: web pages, web forms to include in remote websites, and a RESTful web services API to access remotely from applications. In order…
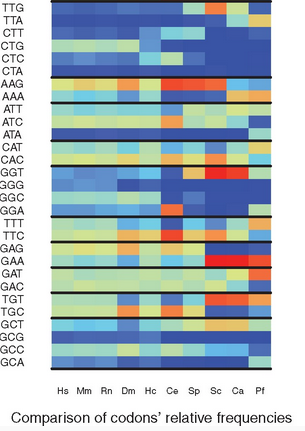
RSCU_RS: Measuring the bias in…
Overview Overview: In the protein coding sequences of a species, the 61 possible codons of the genetic code are not equally distributed. This observation is referred to as the Codon Usage Bias (CUB) of a species. Several measures have been proposed to quantify the CUB using the frequencies of codons in all RNA coding sequences…

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…