EPIK
Precise and scalable evolutionary placement with informative k-mers
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree.
It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS remains valid.
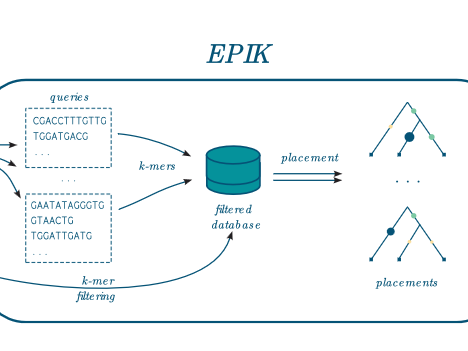
EPIK takes as input a file containing database of phylo-k-mers built with IPK and a set of reads to place on the related phylogeny. It works for nucleotidic and amino-acid query sequences.
EPIK can filter the database to load only the most informative phylo-k-mers, which reduces the memory usage.
EPIK can also run in parallel.
Keywords
Phylogenetic placement, metabarcoding, taxonomic identification, NGS, software

The workflow combining IPK, to precompute the index, and then EPIK, to perform the placement of the reads.
Other tools

TFscope
Characterizing the binding preferences of transcription factors (TFs) in different cell types and conditions is key to understand how they orchestrate gene expression. TFscope is a machine learning approach that identifies sequence features explaining the binding differences observed between two ChIP-seq experiments targeting either the same TF in two conditions or two TFs with similar…

CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared…
RSCU_RS: Measuring the bias in…
Overview Overview: In the protein coding sequences of a species, the 61 possible codons of the genetic code are not equally distributed. This observation is referred to as the Codon Usage Bias (CUB) of a species. Several measures have been proposed to quantify the CUB using the frequencies of codons in all RNA coding sequences…