PEWO: a collection of workflows to benchmark phylogenetic placement
Introduction and context
In the Bioinformatics team of the LIRMM (CNRS & Univ. Montpellier), we develop a series of tools for metagenomics / metabarcoding analysis. Our tools exploit phylo-k-mers (which are k-mers combined with phylogenetic information) computed for an input set of reference sequences and their phylogeny. The phylo-k-mers are computed and indexed with IPK, which stores them in files. Then, using such a phylo-k-mer index (or database):
- EPIK can perform phylogenetic placement of an input set of metabarcoding reads
- SHERPAS identifies reads that are recombinant between different virus strains.
PEWO is a tool to execute, evaluate and compare virtually any tool that does phylogenetic placement of sequencing reads (currently these include Pplacer, EPA, EPA-ng, APPLES, AppSpam, RAPPAS and EPIK).
PEWO: overview

PEWO stands for Phylogenetic placement Evaluation WOrkflows.
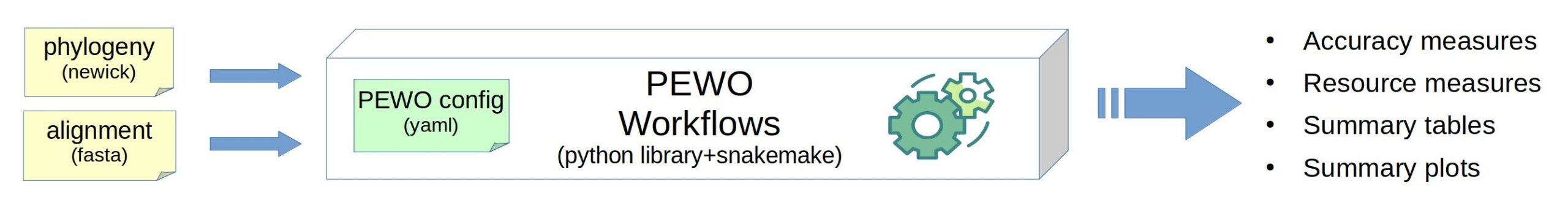
PEWO is a framework to run evaluation and comparison of any tool that performs phylogenetic placement of metagenomic reads. PEWO allows automatic evaluation of precision, of running time and memory usage for several tools on benchmark datasets. It runs the software, collect the results, and prepare graphics for ready-to-use figures. It evaluates all tools in a standard and carefully design procedure: that way you can run a fair comparison, but you can also explore which parameter setting best fits your use-case.
PEWO is freely accessible at https://github.com/phylo42/PEWO and comes with a Wiki, a tutorial, a comprehensive documentation and benchmark datasets.
In 2026, it includes 3 workflows: Pruning-based Accuracy evaluation (PAC), Likelihood-based Accuracy evaluation (LAC), and Resources evaluation (RES).
PEWO is a collaborative effort: test it and make it yours!
Technical overview
PEWO is a framework developped with Snakemake, Python, and Conda (for the management of environments). It already incoporates 7 published, standard tools for phylogenetic placement (see list above). PEWO is flexible and extensible: there is a ligthweight procedure to incorporate a new placement tool. PEWO was published in 2020 and adopted by the community that has already extended it.
License: MIT license.
Publication

Funding
France Génomique [ANR-10-INBS-0009], MNERT fellowship

Other tools

CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared…

TFscope
Characterizing the binding preferences of transcription factors (TFs) in different cell types and conditions is key to understand how they orchestrate gene expression. TFscope is a machine learning approach that identifies sequence features explaining the binding differences observed between two ChIP-seq experiments targeting either the same TF in two conditions or two TFs with similar…
PhyML 3.0
Overview: new algorithms, methods and utilities PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient…