LoRMA: a self correction program for long reads
Overview
LoRMA is an error correction program for long reads, which are sequences obtained using the third generation of sequencing technologies (3GS), either with Oxford Nanopore technology or with Pacific Biosciences technology.
LoRMA is a so-called self-correction software, as opposed to e.g. LoRDEC that is a hybrid error correction tool. This means that LoRMA uses only long read sequencing data and thus does not require short read data.
LoRMA proceeds in two phases.
- It iteratively performs local correction of the long reads using LoRDEC (with one special option). The number of LoRDEC iterations is set by the user.
- It then corrects the long read using long-range sequence similarity, which it detects by clustering similar reads using a heuristic multiple alignment procedure.
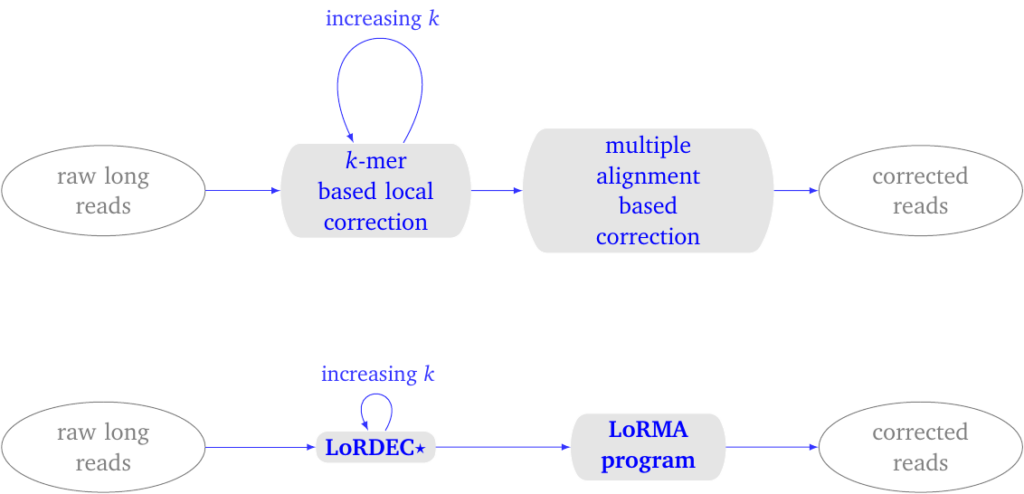
Figure 1: LoRMA process overview. (top) conceptual process. (bottom) pipeline. It uses LoRDEC with a special parameter for self-correction. The entire pipeline can be executed with a single script lorma.sh.
In the first phase, LoRDEC is run with an increasing parameter \(k\), which defines the \(k\) -mers used in the de Bruijn graph. By default, one performs three runs of LoRDEC correction typically with the k-mer sizes 19, 40 and 61 for a yeast data set.
The more iterations, the better the correction, the longer the execution time.
In the second phase, LoRMA process each read in turn. It searches for other reads that share similar regions with the current read. These similar reads are termed friends. An option controls how many friends are sought. It computes a multiple alignment of this subset of reads and uses the consensus sequence to correct the current read.
On this site, we provide the program, easy installation procedures (as a linux package or as a conda package), as well as script for parallel execution on large computing servers.
Publication

Funding
- Current support for maintenance and development of LoRMA: ATGC and IFB
- Supports from Finland for the original research and development of LoRMA: University of Helsinki, SYSCOL project, Helsinki Institute for Information Technology
- Supports from France for the original research and development of LoRMA: LIRMM and Institute of Computational Biology.



Other tools

DExTER
Overview DExTER (Domain Exploration To Explain gene Regulation) is a bioinformatics tool designed to automatically identify genomic regions whose nucleotide composition correlates with gene expression levels. Unlike traditional approaches focusing on short transcription factor binding sites (6-12 bp), DExTER detects Long Regulatory Elements (LREs) that can span tens to hundreds of nucleotides. This makes it…

WAVES
Summary WAVES is a web application dedicated to bioinformatic tool integration. It provides an efficient way to implement a service for any bioinformatic software. Such services are automatically made available in three ways: web pages, web forms to include in remote websites, and a RESTful web services API to access remotely from applications. In order…
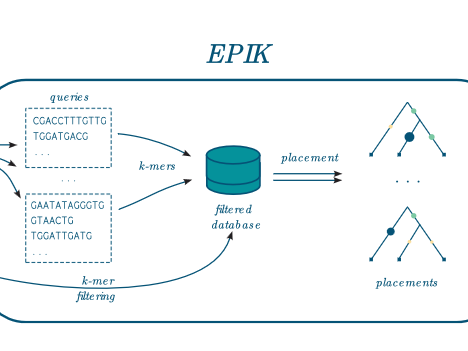
EPIK: Precise and scalable evolutionary…
EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and flexibility. In many aspects the documentation of RAPPAS…