LoRDEC: hybrid correction of long reads
Overview
In a nutshell, LoRDEC is a program for error correcting long sequencing reads using short reads. It implements a hybrid correction approach. It uses little memory and is very efficient. Most importantly it scales up to process very large data sets. It can be applied to long reads obtained with either Pacific Biosciences SMRT sequencing (SMRT = Single Molecule Real Time) or with Oxford Nanopore MINion technology.
Why is LoRDEC algorithm different?
- It is efficient and can process large read data sets, included from eukaryotic or vertebrate species, on a usual computing server, and even works on desktop/laptop computers.
- It adopts a novel graph based approach: it builds a succinct De Bruijn Graph (DBG) representing the short reads, and seeks a corrective sequence for each erroneous region of a long read by traversing chosen paths in the graph.
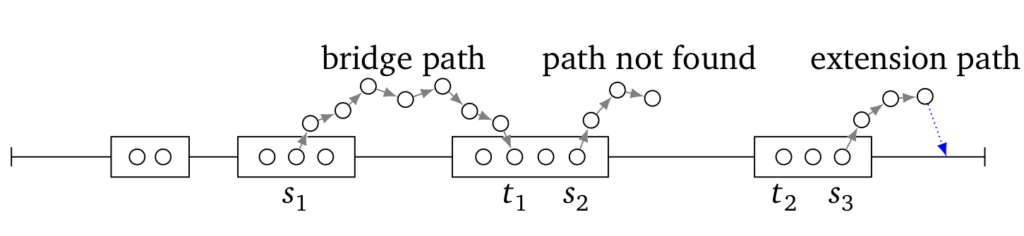
For the sake of efficiency, LoRDEC analyzes reads through their k-mers. If a k-mer is error-free, we call it solid (circles).
Long read is viewed by LoRDEC as a series of blocks of error-free k-mers (drawn as rectangles), separated by regions of erroneous k-mers (horizontal line). To bridge erroneous regions, LoRDEC finds path in a De Bruijn graph of the short reads (shown as directed paths of circles outside the horizontal line). Such correction paths are shown as directed paths of circles outside the horizontal line.
More infos
LoRDEC is a bioinformatics software published and initially developed in collaboration with Leena Salmela of University of Helsinki (Finland).
LoRDEC processes data coming from high throughput sequencing machines of the second and third generations. These data are called sequencing reads, or simply reads for short. Technically speaking it processes short reads and long reads to correct errors in the long reads.
Third generation DNA sequencing technologies yield long, but erroneous sequencing reads, while second generation technologies yield short read with low error rate. Hence, the need for correcting the long reads.
LoRDEC has been accepted by the Elixir Service Delivery Plan in 2019, and since then LoRDEC is registered in the ELIXIR bio.tools database under this entry), and documented following the EDAM ontology.
LoRDEC has been used in large genome and transcriptome projects by numerous genomic institutes over the world. Some infos about the first hundred such projects can be found at https://www.lirmm.fr/~rivals/lordec/.
High Performance Computing
We also provide scripts for a parallel use of LoRDEC on High Performance Computing servers. If you want to correct long reads but lack short reads, then you can look at LoRMA, which is non hybrid error correction tool for long reads.
- More information at including FAQ at https://www.lirmm.fr/~rivals/lordec/
- Official releases page at our gitlab server: https://gite.lirmm.fr/lordec/lordec-releases/wikis/home
NB: Elixir is a European organisation that coordinates life science resources from across Europe, and the Institut Français de Bioinformatique is the French node of Elixir.
Funding
This work was supported by
- Academy of Finland [grant number 267591]
- ANR Colib’read (ANR-12-BS02-0008)
- Défi MASTODONS SePhHaDe from CNRS
- Labex NumEV

Other tools

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…
PhyML 3.0
Overview: new algorithms, methods and utilities PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient…

DExTER
Overview DExTER (Domain Exploration To Explain gene Regulation) is a bioinformatics tool designed to automatically identify genomic regions whose nucleotide composition correlates with gene expression levels. Unlike traditional approaches focusing on short transcription factor binding sites (6-12 bp), DExTER detects Long Regulatory Elements (LREs) that can span tens to hundreds of nucleotides. This makes it…