AQUAPONY
AquaPony: interactive visualization of phylogeographic scenarios
AquaPony is a web application designed to explore and interpret evolutionary scenarios on annotated phylogenetic trees (for example, ancestral geographic states). It was built to make uncertainty in ancestral reconstructions easier to understand and communicate.
Why AquaPony?
In phylogeography, several scenarios can be nearly as plausible as the best one. AquaPony helps users go beyond a single “best” reconstruction by interactively examining alternative scenarios under user-defined uncertainty thresholds.
Key features
- Interactive visualization of the full tree and selected subtrees.
- Explicit display of uncertainty in ancestral state assignments.
- Exploration of alternative phylogeographic scenarios along branches.
- Instant visual updates, including for large trees.
- Export-ready figures for reports and publications.
Use cases
AquaPony is relevant to infectious disease research, ecology, agronomy, and more broadly to any study involving spatiotemporal evolutionary inference from annotated phylogenies.
Access the tool
Open AquaPony here: https://website.atgc-montpellier.fr/aquapony/aquapony.html
Scientific reference
Cazaux B., Castel G., Rivals E. (2019). AQUAPONY: visualization and interpretation of phylogeographic information on phylogenetic trees. Bioinformatics, 35(17), 3163–3165. DOI: 10.1093/bioinformatics/btz011

Other tools

FastME 2.0
FastME is a software package for the fast and accurate inference of phylogenetic trees from distance matrices. It implements algorithms based on the Balanced Minimum Evolution (BME) principle, a distance-based criterion closely related to the Neighbor Joining (NJ) method. The goal of the BME framework is to identify the phylogenetic tree that minimizes the total…
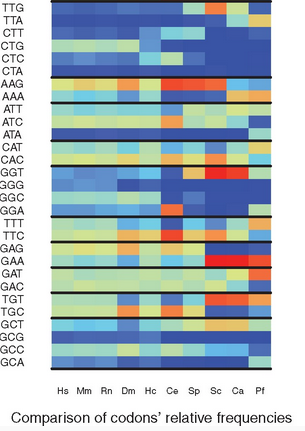
RSCU_RS: Measuring the bias in…
Overview Overview: In the protein coding sequences of a species, the 61 possible codons of the genetic code are not equally distributed. This observation is referred to as the Codon Usage Bias (CUB) of a species. Several measures have been proposed to quantify the CUB using the frequencies of codons in all RNA coding sequences…
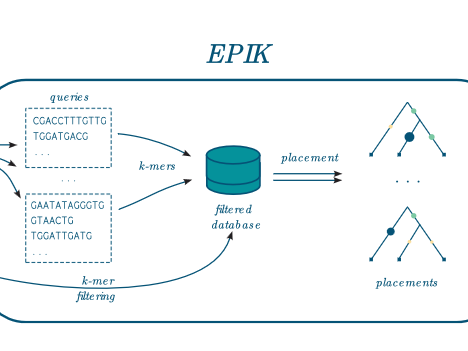
EPIK
Precise and scalable evolutionary placement with informative k-mers EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and…