TIDE
What is TIDE?
TIDE is the orchestration platform behind ATGC online bioinformatics services. It provides a unified web interface and a public REST API to submit analyses, monitor jobs, and retrieve results across a growing catalog of phylogenetic and sequence-analysis tools.
TIDE is a modernized redesign of WAVES, an older tool previously developped by ATGC and now abandoned. It keeps the same general objective of exposing scientific tools as online services, while moving to a more modern, maintainable, and extensible platform.
TIDE on the ATGC website
TIDE is the engine that powers the online execution proposed on the dedicated pages of several ATGC tools. When you submit a job from one of these tool pages, it is TIDE that manages the service description, input validation, job submission, execution tracking, and result retrieval behind the scenes.
What TIDE provides
- A public REST API for programmatic submissions and monitoring.
- A web interface for interactive use.
- Reproducible execution of tools packaged as portable runtime environments.
- Service-specific forms and structured input schemas.
- Downloadable recipes and reference resources for selected services.
- Job tracking, status updates, and result retrieval.
Current usage policy
At the moment, TIDE is limited to 5 jobs per user per hour. This helps us keep the platform stable while the service catalog is growing.
If you need access to more resources or expect heavier usage, please contact us. We can discuss specific needs depending on the project, the service, and the computational load involved.
Why it matters
TIDE helps bridge the gap between command-line bioinformatics tools and production-ready scientific services. It allows ATGC to expose tools through a stable interface while keeping execution traceable, reproducible, and suitable for both end users and automated pipelines.
Current status and roadmap
For now, TIDE is primarily developed to address the specific needs of the ATGC platform and its service ecosystem.
A more general, publicly distributable version is planned for future release. If you are interested in deploying or evaluating TIDE outside the current ATGC context, please contact us.
Access points
Typical usage
- Discover available services through the API.
- Inspect the submission schema of a service.
- Submit a job from the web interface or from the command line.
- Monitor execution through JSON status checks or live event streaming.
- Retrieve final outputs and integrate them into downstream analyses.
For developers and pipeline users
TIDE is especially useful when you need to integrate ATGC services into scripts, workflows, or external platforms. The public API exposes machine-readable service descriptions, structured submission endpoints, and result-tracking endpoints suitable for automation.
Read the documentation page for API entry points, recommended workflow, and ready-to-use command-line examples.

Other tools

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…
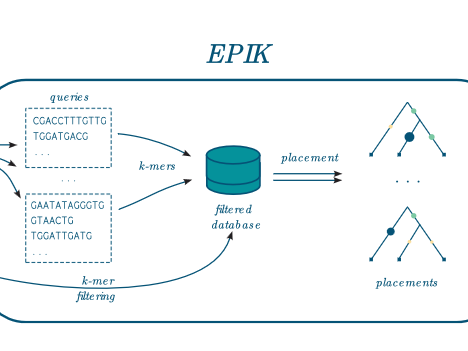
EPIK
Precise and scalable evolutionary placement with informative k-mers EPIK is a program dedicated to « Phylogenetic Placement » (PP) of metagenomic or metabarcoding reads on a reference tree. It is similar in spirit and technically the successor of RAPPAS (Linard et al. 2020). EPIK achieves identical or slightly better accuracy than RAPPAS and outperforms it in speed and…
PhyML 3.0
Overview: new algorithms, methods and utilities PhyML is a software package that uses modern statistical approaches to build phylogenetic trees from the analysis of alignments of nucleotide or amino acid sequences. The main tool in this package builds phylogenies under the maximum likelihood criterion. It implements a large number of substitution models coupled to efficient…