RSCU_RS: Measuring the bias in codon usage from ribosomal activity
Overview
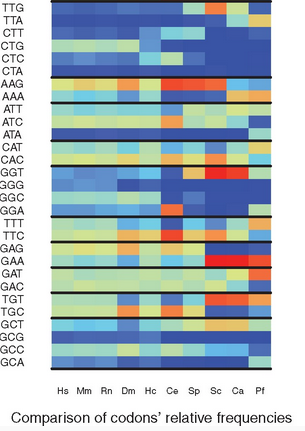
Overview: In the protein coding sequences of a species, the 61 possible codons of the genetic code are not equally distributed. This observation is referred to as the Codon Usage Bias (CUB) of a species. Several measures have been proposed to quantify the CUB using the frequencies of codons in all RNA coding sequences (or at least a representative subset of these). This yields a static analysis since sequences change slowly over time. But in living cells the translation varies over time, since the ribosome, the molecular machine that performs translation, has a role in the selection of which RNA sequences are indeed translated. The precise location of ribosomes translating RNA can be monitor by Ribo-seq (aka ribosome profiling), a high throughput sequencing assay that captures the portions of RNA located inside the ribosomes. Thus, the sequencing reads produced by Ribo-seq in a given condition give us access to which codons ribosomes are translating.
We proposed to measure the codon usage bias in a transcriptome wide manner using Ribo-seq sequencing data. This delivers a dynamic and precise estimation of Codon Usage Bias, since it integrates the location of ribosomes during translation. The codon usage bias can be computed classicaly for a given species, for a subset of genes, but also for any given condition for which Ribo-seq data is available. The CUB can thus be compared across species, across subsets of genes, or across conditions.
For this, we develop a stand-alone software, called RSCU_RS, and report experiments in estimating and comparing CUB across species in a journal article.
Reference
Journal article
Ribo-seq enlightens Codon Usage Bias
D. Paulet, A. David, E. Rivals
DNA Research, dsw062. https://doi.org/10.1093/dnares/dsw062, 2017.

Documentation and ressources
A documentation about the software RSCU_RS is available at
https://www.lirmm.fr/~rivals/rscu/
Funding
Fondation pour la Recherche Médicale, grant DBI20131228574.
ANR grant (ANR-11-BINF-0002), Institut de Biologie Computationnelle (IBC).
Other tools

AQUAPONY
AquaPony: interactive visualization of phylogeographic scenarios AquaPony is a web application designed to explore and interpret evolutionary scenarios on annotated phylogenetic trees (for example, ancestral geographic states). It was built to make uncertainty in ancestral reconstructions easier to understand and communicate. Why AquaPony? In phylogeography, several scenarios can be nearly as plausible as the best…

dipwmsearch
Protein binding sites in DNA or RNA sequences are modeled by probabilistic motifs. A Position Weight Matrix (PWM) is a simple, powerful, and widely used representation of such motifs. Because PWMs assume that sequence positions are independent of eachother (which is too restrictive for some binding or interaction sites), a generalisation of PWMs, termed di-nucleotidic…

CompPhy
CompPhy: a web-based collaborative platform for comparing phylogenies CompPhy is a web platform dedicated to the collaborative handling of phylogenetic trees. Users can freely manage collections of trees and communicate on a common project. By collaborative, we mean that several users connected to the same project can manipulation at the same time trees from shared…